Recombinant Human Alsin (ALS2), partial
-
中文名称:Recombinant Human Alsin(ALS2) ,partial
-
货号:CSB-YP857009HU
-
规格:
-
来源:Yeast
-
其他:
-
中文名称:Recombinant Human Alsin(ALS2) ,partial
-
货号:CSB-EP857009HU
-
规格:
-
来源:E.coli
-
其他:
-
中文名称:Recombinant Human Alsin(ALS2) ,partial
-
货号:CSB-EP857009HU-B
-
规格:
-
来源:E.coli
-
共轭:Avi-tag Biotinylated
E. coli biotin ligase (BirA) is highly specific in covalently attaching biotin to the 15 amino acid AviTag peptide. This recombinant protein was biotinylated in vivo by AviTag-BirA technology, which method is BriA catalyzes amide linkage between the biotin and the specific lysine of the AviTag.
-
其他:
-
中文名称:Recombinant Human Alsin(ALS2) ,partial
-
货号:CSB-BP857009HU
-
规格:
-
来源:Baculovirus
-
其他:
-
中文名称:Recombinant Human Alsin(ALS2) ,partial
-
货号:CSB-MP857009HU
-
规格:
-
来源:Mammalian cell
-
其他:
产品详情
-
纯度:>85% (SDS-PAGE)
-
基因名:ALS2
-
Uniprot No.:
-
别名:ALS 2; ALS2; ALS2_HUMAN; ALS2CR6; Alsin; ALSJ; Amyotrophic lateral sclerosis 2 (juvenile); Amyotrophic lateral sclerosis 2 (juvenile) chromosome region candidate 6; Amyotrophic lateral sclerosis 2 chromosomal region candidate gene 6 protein; Amyotrophic lateral sclerosis 2 protein; Amyotrophic lateral sclerosis protein 2; FLJ31851; IAHSP; KIAA1563; MGC87187; PLSJ
-
种属:Homo sapiens (Human)
-
蛋白长度:Partial
-
蛋白标签:Tag type will be determined during the manufacturing process.
The tag type will be determined during production process. If you have specified tag type, please tell us and we will develop the specified tag preferentially. -
产品提供形式:Lyophilized powder
Note: We will preferentially ship the format that we have in stock, however, if you have any special requirement for the format, please remark your requirement when placing the order, we will prepare according to your demand. -
复溶:We recommend that this vial be briefly centrifuged prior to opening to bring the contents to the bottom. Please reconstitute protein in deionized sterile water to a concentration of 0.1-1.0 mg/mL.We recommend to add 5-50% of glycerol (final concentration) and aliquot for long-term storage at -20℃/-80℃. Our default final concentration of glycerol is 50%. Customers could use it as reference.
-
储存条件:Store at -20°C/-80°C upon receipt, aliquoting is necessary for mutiple use. Avoid repeated freeze-thaw cycles.
-
保质期:The shelf life is related to many factors, storage state, buffer ingredients, storage temperature and the stability of the protein itself.
Generally, the shelf life of liquid form is 6 months at -20°C/-80°C. The shelf life of lyophilized form is 12 months at -20°C/-80°C. -
货期:Delivery time may differ from different purchasing way or location, please kindly consult your local distributors for specific delivery time.Note: All of our proteins are default shipped with normal blue ice packs, if you request to ship with dry ice, please communicate with us in advance and extra fees will be charged.
-
注意事项:Repeated freezing and thawing is not recommended. Store working aliquots at 4°C for up to one week.
-
Datasheet :Please contact us to get it.
产品评价

相关产品
靶点详情
-
功能:May act as a GTPase regulator. Controls survival and growth of spinal motoneurons.
-
基因功能参考文献:
- This study identified a novel ALS2 pathogenic founder variant in Iran that further adds to the allelic heterogeneity of infantile-onset ascending hereditary spastic paralysis. PMID: 30128655
- Nonsense mutation in ALS2 gene is associated with severe and progressive infantile onset of spastic paralysis. PMID: 28502191
- We established a genetic diagnosis in six families with autosomal recessive HSP (SPG11 in three families and TFG/SPG57, SACS and ALS2 in one family each). A heterozygous mutation in a gene involved in an autosomal dominant HSP (ATL1/SPG3A) was also identified in one additional family PMID: 27601211
- This study identified two novel ALS2 mutations in two Pakistani families with infantile-onset ascending hereditary spastic paraplegia cosegregating with the disease. PMID: 26751646
- novel compound heterozygous ALS2 deletion mutations were identified in two siblings with infantile ascending hereditary spastic paraplegia. PMID: 25433428
- We identified a novel homozygous splice-site mutation (c.3512+1G>A) in the ALS2 gene (NM_020919.3) encoding alsin that segregated with the disease in this family PMID: 25474699
- Data indicate a splice-site mutation of the amyotrophic lateral sclerosis 2 (juvenile) protein (ALS2) in four children of a consanguineous family with infantile-onset ascending hereditary spastic paraplegia. PMID: 24704789
- The ALS2 gene should be screened for mutations in patients who present with generalized dystonia and cerebellar signs. PMID: 24562058
- The ALS2 mutation c.2761C>T leading to infantile-onset hereditary spastic paraplegia resides in the pleckstrin domain, which is involved in the overall neuronal development or maintenance. PMID: 24315819
- ALS2 sequencing revealed two heterozygous mutations: the missense variant c.299 G>T, leading to the replacement of a serine with an isoleucine (p.S100I), and the splicing variant c.2580-2 A>G in brothers with juvenile amyotrophic lateral sclerosis. PMID: 23282280
- these results suggest that Als2 is a binding partner of Uxt and Als2/Uxt interaction could be important for the activation of Nf-kappaB pathway. PMID: 21907703
- causative genes for familial amyotrophic lateral sclerosis PMID: 12138710
- Infantile-onset ascending hereditary spastic paralysis is associated with mutations in the alsin gene. PMID: 12145748
- 16 patients from 11 unrelated families were studied with a phenotype of infantile ascending hereditary spastic paralysis (IAHSP); Alsin mutations were found in 4 of the 10 families, whereas haplotype analysis excluded the ALS2 locus in one family PMID: 12601111
- Perturbation of endosomal dynamics caused by loss of ALS2 rab5GEF activity might underlie neuronal dysfunction and degeneration. PMID: 12837691
- deletion mutations in ALS2 gene detected in ALS2 patients seem to be uncommon in Japanese AR-ALS, and that SNPs in uncoding regions might possibly be relevant to predisposition to ALS. PMID: 12866199
- A nonsense mutation in alsin was found in infantile spastic paraplegia. Full-length alsin is probably required for the proper development and/or functioning of upper motor neurons. PMID: 12919135
- Mutations in the ALS2 gene linked to early-onset motor neuron disease uniformly produce loss of activity through decreased protein stability of this endosomal protein. PMID: 14668431
- Mutations of ALS2 are not a common cause of ALS. PMID: 14676054
- Expression of alsin LF, but not alsin short form, protected motor neuronal cells from toxicity induced by mutants of the Cu/Zn-superoxide dismutase (SOD1) gene, which cause autosomal dominant ALS PMID: 14970233
- oligomerization of the ALS2 protein is one of the fundamental features for its physiological function involving endosome dynamics in vivo PMID: 15247254
- A peptide derived from the ALS2 protein is selectively localized to the somatodendritic compartment of motor neurons in human spinal cord. PMID: 15371724
- These results suggest that amyotrophic lateral sclerosis 2 C-terminal like (ALS2CL), a novel ALS2 homologue, modulates Rab5-mediated endosome dynamics in HeLa cells. PMID: 15388334
- Rac1, PI3 kinase, and Akt3 have roles in an anti-apoptotic pathway triggered by ALS2 that antagonizes SOD1 mutant-induced motoneuronal cell death PMID: 15579468
- ALS2/Alsin has a role in regulating Rac-PAK signaling and neurite outgrowth PMID: 16049005
- colocalization of Alsin with the centrosomal markers gamma-tubulin and A kinase anchoring protein. PMID: 16085057
- ALS2 mutations are not implicated in the pathogenesis of adult-onset primary lateral sclerosis. PMID: 17698795
- Autosomal recessive mutations in the ALS2 gene lead to a clinical spectrum of motor dysfunction including juvenile onset amyotrophic lateral sclerosis. [REVIEW] PMID: 17955197
- mutations in ALS2 also need to be considered in patients from northwestern Europe with early-onset spastic paralysis and amyotrophic or primary lateral sclerosis. PMID: 18523452
- Results suggest at least four recombination events in the ALS2 gene during maternal meiosis followed by a meiosis I error and postzygotic trisomy rescue or gamete complementation, in a patient with infantile-onset ascending spastic paralysis. PMID: 18810511
- A structural model for the N-terminal 690-residue region of alsin through comparative modelling based on regulator of chromosome condensation 1 was created. PMID: 19023603
- This novel ALS2 splice-site mutation is causing the loss of exon 18 in the transcript which results in a frameshift after exon 17. PMID: 19122027
显示更多
收起更多
-
相关疾病:Amyotrophic lateral sclerosis 2 (ALS2); Juvenile primary lateral sclerosis (JPLS); Infantile-onset ascending spastic paralysis (IAHSP)
-
数据库链接:
Most popular with customers
-

Recombinant Human CD40 ligand (CD40LG), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
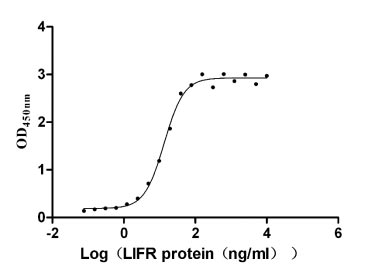
Recombinant Human Leukemia inhibitory factor receptor (LIFR), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
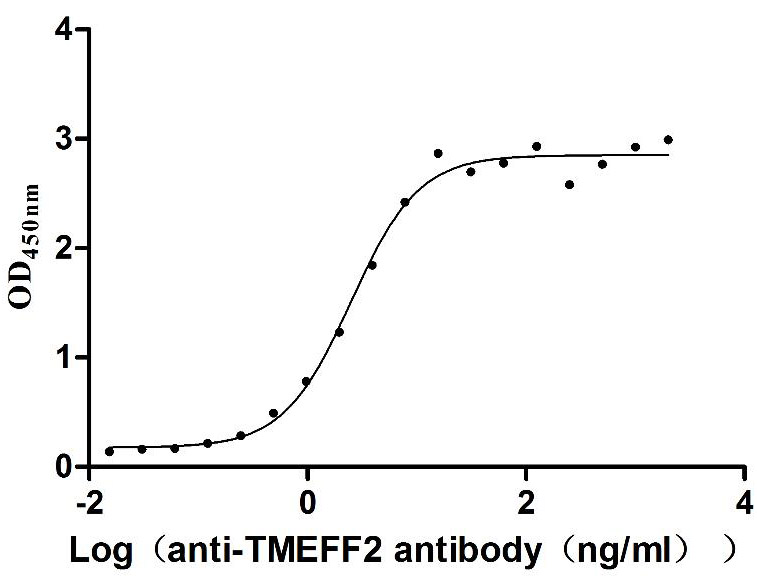
Recombinant Human Tomoregulin-2 (TMEFF2), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
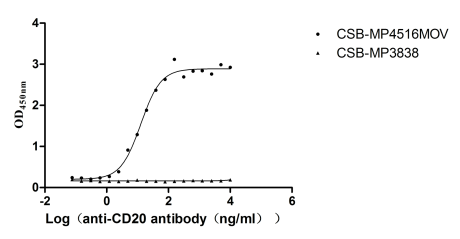
Recombinant Macaca fascicularis Membrane spanning 4-domains A1 (MS4A1)-VLPs (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
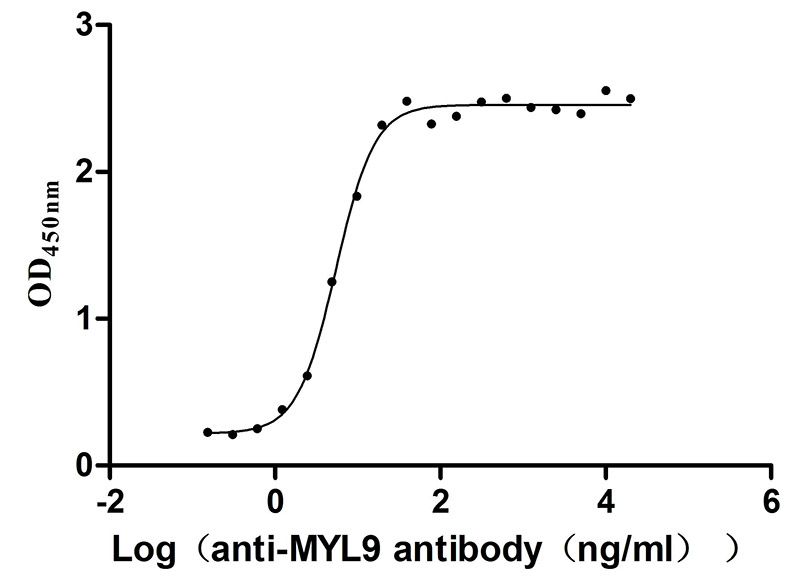
Recombinant Human Myosin regulatory light polypeptide 9 (MYL9) (Active)
Express system: Yeast
Species: Homo sapiens (Human)
-
Recombinant Human Killer cell immunoglobulin-like receptor 3DL2 (KIR3DL2), partial (Active)
Express system: Mammalian cell
Species: Homo sapiens (Human)
-
Recombinant Macaca fascicularis Transmembrane 4 L6 family member 1 (TM4SF1)-VLPs (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)
-
Recombinant Macaca fascicularis Dipeptidase 3(DPEP3) (Active)
Express system: Mammalian cell
Species: Macaca fascicularis (Crab-eating macaque) (Cynomolgus monkey)









